
Abstract
Data-independent acquisition (DIA) is an emerging technology for quantitative proteomic analysis of large cohorts of samples. However, sample-specific spectral libraries built by data-dependent acquisition (DDA) experiments are required prior to DIA analysis, which is time-consuming and limits the identification/quantification by DIA to the peptides identified by DDA. Herein, we propose DeepDIA, a deep learning-based approach to generate in silico spectral libraries for DIA analysis. We demonstrate that the quality of in silico libraries predicted by instrument-specific models using DeepDIA is comparable to that of experimental libraries, and outperforms libraries generated by global models. With peptide detectability prediction, in silico libraries can be built directly from protein sequence databases. We further illustrate that DeepDIA can break through the limitation of DDA on peptide/protein detection, and enhance DIA analysis on human serum samples compared to the state-of-the-art protocol using a DDA library. We expect this work expanding the toolbox for DIA proteomics.
Similar content being viewed by others

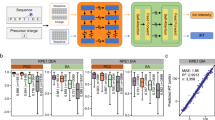

Introduction
With the ability to identify and precisely quantify thousands of proteins from complex samples, liquid chromatography (LC)-tandem mass spectrometry (MS/MS) has been the most widely used tool for proteomic studies over the past decadesUniProt. The mouse data were searched against the SwissProt Mus musculus database (access date 2019-02, 17,006 entries). Q-value cutoff on precursor and protein level was applied 1%. Other parameters were default values.
Training and validation of the deep neural networks
HCD MS/MS spectra of peptide precursors were collected from the HeLa1 (17 runs), HeLa2 (12 runs), Mouse1 (23 runs), and Mouse2 (15 runs) DDA data (Supplementary Table Anaconda distribution version 4.2.0) using Keras (version 2.2.4) with TensorFlow (version 1.11.0) backend. Data preprocessing and visualization were conducted with R (version 3.5.1). Running time for model training is described in Supplementary Note Protein Digestion Simulator (version 2.2.6794). For in silico libraries without detectability filtering, Trypsin and Trypsin/P were set as the digestion enzyme with no missed cleavages, respectively, and the results were combined. For libraries with detectability filtering, Trypsin/P was set as the digestion enzyme with missed cleavages ≤2. Only peptides with length from 7 to 50 amino acids with mass ≤ 6000 Da were kept.
DIA data analysis
Raw data of DIA were processed and analyzed by Spectronaut. Retention time prediction type was set to dynamic iRT. Data extraction was determined by Spectronaut based on the extensive mass calibration. Decoy generation was set to mutated. Interference correction on MS2 level was enabled. Peptide and protein level Q-value cutoff was set to 1%. For mixed proteome samples, SwissProt H. sapiens isoform database (access date 2018-06, 42,356 entries), UniProt Proteome C. elegans isoform database (access date 2019-03, 28,302 entries), SwissProt S. cerevisiae (strain ATCC 204508 / S288c) database (access date 2019-03, 6,721 entries) and SwissProt E. coli (strain K12) database (access date 2019-03, 4,480 entries) were used as protein sequence databases. For other datasets, protein database was set the same as those used in DDA searching.
For large spectral libraries, machine learning was performed across experiments, and protein groups with single hit (i.e. only one stripped peptide sequence) in each run were excluded. An entrapment strategy37 was used to compare false positive identification rates under the given Q-value. An entrapment library was built using proteins from other organisms with roughly equivalent size to the organism specific library (see Supplementary Table 2 for details). The organism specific library and the entrapment library were merged and used as the target library. Identification results were filtered by 1% Q-value by a target-decoy approach implemented by Spectronaut. The generation of decoy was on the whole target library including entrapment. As we introduced the entrapment entries in the target database, the entrapment hits in filtered target hits were considered as false positive results. Thus, we used entrapment percentage (percentage of the number of entrapment hits to the target hits) to compare the false positive rates relatively. It should be noted that the true error rate is higher than the entrapment percentage.
Peptide and protein reports were exported as CSV files, and subsequent statistic and visualization were performed with R scripts.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this Article.
Data availability
All raw mass spectrometry data, spectral libraries and search results are publicly available at the ProteomeXchange Consortium. Raw data of HeLa, HEK-293, mouse and mixed proteome samples are available with the dataset identifier PXD005573, PXD006932, PXD004452, and PXD009875 (see Supplementary Table PXD014108 and IPX0001628000. The source data underlying Figs. 2c-d and 3b-d, as well as Supplementary Fig. 1, 2, 3b-c, 4b-c, 5b-c, 7, 8c and 10b are provided as a Source Data file. All other data are available from the corresponding author on reasonable request.
Code availability
DeepDIA is open source and freely available on GitHub.
References
Aebersold, R. & Mann, M. Mass spectrometry-based proteomics. Nature 422, 198–207 (2003).
Domon, B. & Aebersold, R. Mass spectrometry and protein analysis. Science 312, 212–217 (2006).
Gillet, L. C. et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 11, O111.016717 (2012).
Ting, Y. S. et al. Peptide-centric proteome analysis: an alternative strategy for the analysis of tandem mass spectrometry data. Mol. Cell. Proteom. 14, 2301–2307 (2015).
Tsou, C.-C. et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264 (2015).
Li, Y. et al. Group-DIA: analyzing multiple data-independent acquisition mass spectrometry data files. Nat. Methods 12, 1105–1106 (2015).
Röst, H. L. et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 (2014).
Wang, J. et al. MSPLIT-DIA: sensitive peptide identification for data-independent acquisition. Nat. Methods 12, 1106–1108 (2015).
Navarro, P. et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 34, 1130–1136 (2016).
Ludwig, C. et al. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 14, e8126 (2018).
Moruz, L. & Käll, L. Peptide retention time prediction. Mass Spectrom. Rev. 36, 615–623 (2017).
Escher, C. et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 (2012).
Gorshkov, A. V. et al. Liquid chromatography at critical conditions: comprehensive approach to sequence-dependent retention time prediction. Anal. Chem. 78, 7770–7777 (2006).
Meek, J. L. Prediction of peptide retention times in high-pressure liquid chromatography on the basis of amino acid composition. Proc. Natl Acad. Sci. USA 77, 1632–1636 (1980).
Krokhin, O. V. et al. An improved model for prediction of retention times of tryptic peptides in ion pair reversed-phase HPLC. Mol. Cell. Proteom. 3, 908–919 (2004).
Bereman, M. S., MacLean, B., Tomazela, D. M., Liebler, D. C. & MacCoss, M. J. The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 12, 1134–1141 (2012).
Petritis, K. et al. Use of artificial neural networks for the accurate prediction of peptide liquid chromatography elution times in proteome analyses. Anal. Chem. 75, 1039–1048 (2003).
Moruz, L., Tomazela, D. & Käll, L. Training, selection, and robust calibration of retention time models for targeted proteomics. J. Proteome Res. 9, 5209–5216 (2010).
Reimer, J., Spicer, V. & Krokhin, O. V. Application of modern reversed-phase peptide retention prediction algorithms to the houghten and degraw dataset: peptide helicity and its effect on prediction accuracy. J. Chromatogr. A 1256, 160–168 (2012).
Zhang, Z. Prediction of low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 3908–3922 (2004).
Sun, S. et al. MS-simulator: predicting y-ion intensities for peptides with two charges based on the intensity ratio of neighboring ions. J. Proteome Res. 11, 4509–4516 (2012).
Arnold, R. J., Jayasankar, N., Aggarwal, D., Tang, H. & Radivojac, P. A machine learning approach to predicting peptide fragmentation spectra. Pac. Symp. Biocomput. 11, 219–230 (2006).
Degroeve, S., Maddelein, D. & Martens, L. MS2PIP prediction server: Compute and visualize MS2 peak intensity predictions for CID and HCD fragmentation. Nucleic Acids Res. 43, W326–W330 (2015).
Li, S., Arnold, R. J., Tang, H. & Radivojac, P. On the accuracy and limits of peptide fragmentation spectrum prediction. Anal. Chem. 83, 790–796 (2011).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Zhou, X.-X. et al. pDeep: predicting MS/MS spectra of peptides with deep learning. Anal. Chem. 89, 12690–12697 (2017).
Gessulat, S. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518 (2019).
Tiwary, S. et al. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 16, 519–525 (2019).
Tran, N. H. et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 16, 63–66 (2019).
Bruderer, R. et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen treated 3D liver microtissues. Mol. Cell. Proteom. 14, 1400–1410 (2015).
Bruderer, R. et al. Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteom. 16, 2296–2309 (2017).
Kelstrup, C. D. et al. Performance evaluation of the Q Exactive HF-X for shotgun proteomics. J. Proteome Res. 17, 727–738 (2018).
Wan, K. X., Vidavsky, I. & Gross, M. L. Comparing similar spectra: from similarity index to spectral contrast angle. J. Am. Soc. Mass Spectrom. 13, 85–88 (2002).
Bekker-Jensen, D. B. et al. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 4, 587–599 (2017).
Zhang, Y., Wen, Z., Washburn, M. P. & Florens, L. Evaluating chromatographic approaches for the quantitative analysis of a human proteome on orbitrap-based mass spectrometry systems. J. Proteome Res. 18, 1857–1869 (2019).
Rosenberger, G. et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data 1, 140031 (2014).
Feng, X.-D. et al. Using the entrapment sequence method as a standard to evaluate key steps of proteomics data analysis process. BMC Genomics 18, 143 (2017).
Bruderer, R., Bernhardt, O. M., Gandhi, T. & Reiter, L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics 16, 2246–2256 (2016).
Rosenberger, G. et al. Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat. Methods 14, 921–927 (2017).
Issaq, H. J., Xiao, Z. & Veenstra, T. D. Serum and plasma proteomics. Chem. Rev. 107, 3601–3620 (2007).
Addona, T. A. et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 29, 635–643 (2011).
Hortin, G. L., Sviridov, D. & Anderson, N. L. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin. Chem. 54, 1608–1616 (2008).
Wichmann, C. et al. MaxQuant.Live enables global targeting of more than 25,000 peptides. Mol. Cell. Proteom. 18, 982–994 (2019).
Lee, H., Grosse, R., Ranganath, R. & Ng, A. Y. Convolutional deep belief networks for scalable unsupervised learning of hierarchical representations. In Proc. 26th Annual International Conference on Machine Learning 609–616 (ACM, Montreal, Quebec, Canada, 2009).
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput. 9, 1735–1780 (1997).
Srivastava, N., Hinton, G. E., Krizhevsky, A., Sutskever, I. & Salakhutdinov, R. R. Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15, 1929–1958 (2014).
Glorot, X., Bordes, A. & Bengio, Y. Deep sparse rectifier neural networks. In Proc. Fourteenth International Conference on Artificial Intelligence and Statistics 315–323 (PMLR, Fort Lauderdale, FL, USA, 2011).
Kingma, D. P. & Ba, J. Adam: A method for stochastic optimization. Preprint at https://ui.adsabs.harvard.edu/abs/2014arXiv1412.6980K (2014).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, 81671849, 31800691, 21874026), Science and Technology Commission of Shanghai Municipality (18441901000), Ministry of Science and Technology of China (MOST, 2016YFE0132400), the Special Project on Precision Medicine under the National Key R&D Program (2017YFC0906600), and the National Key Research and Development Program of China (2017YFA0505100). Human serum samples were collected under the consent of the donors. The protocol of blood collection, processing and MS analysis was approved by the Medical Ethics Committee of Shanghai Stomatological Hospital affiliated to Fudan University ([2016]0001), and complied with all relevant laws and regulations of China.
Author information
Authors and Affiliations
Contributions
L.Q. supervised all aspects of the study. Y.Y. did all the coding work and data analysis. X.L. and C.S. designed and conducted wet-lab experiments. P.Y. helped design the MS/MS prediction strategy. Y.L. helped design DIA data analysis framework. Y.Y. and L.Q. wrote the paper. All authors were involved in the design of this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Ming Li, and other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Yang, Y., Liu, X., Shen, C. et al. In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics. Nat Commun 11, 146 (2020). https://doi.org/10.1038/s41467-019-13866-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13866-z
