信息中心



在多相催化中👣,催化劑顆粒的尺寸和形貌會極大的影響催化活性,因此其結構-活性關系是多相催化研究中的核心問題之一。催化劑活性由表面電子結構決定,因而多數“結構-活性”研究都集中在構建“結構-能量”關系這一部分。從能量關系出發🔍,通過求解微動力學模型,可以得到反應速率等動力學信息。對於多相催化中常見的納米催化劑😐,目前最有效的微動力學模型是動力學蒙特卡洛模擬(KMC),但受製於計算量,其應用通常被局限於小粒徑的納米顆粒(<5nm)研究中🧑🦳。如果能夠跨越DFT計算和KMC模擬步驟,直接構建催化劑表面活性位點的“結構-活性”關系,將大幅提升催化劑設計與篩選的效率🌠,有力地擴展第一性原理在多相催化研究中的應用範圍。
https://xdft.fudan.edu.cn)發現,對於加氫/脫氫步驟控製的反應體系,由於氫在金屬表面的吸附能對吸附位點不敏感,可以建立單個吸附位點與反應活性的對應關系,且納米催化顆粒的活性可以由各表面位點的活性相加得到🏄🏼♀️🎋。基於單位點模型,徐昕教授課題組對Cu納米顆粒催化的水汽變換反應進行了研究🤘🏿,並對大粒徑Cu納米顆粒(500-1000nm)的活性進行了預測,其預測表觀活化能與實驗值偏差小於1.0 kcal/mol。研究結果還表明,即使對於大粒徑納米顆粒,其催化活性也不能簡單等價於面位的活性🧝🏻♂️🫰,例如對於877nm的正方體顆粒🤦🏽,其棱位對活性仍有較大貢獻👩🏻🏫。這一研究成果以“Structure-Reactivity Relationship for Nano-Catalysts in the Hydrogenation /Dehydrogenation Controlled Reaction Systems”為題發表在《德國應用化學》雜誌(Angew. Chem. Int. Ed. 2021, 60, 26342–26345)。課題組成員申同昊博士與博士生楊宇琦為共同一作👋。該工作得到了國家自然科學基金(Grant 21688102)及國家重點研發計劃(Grant 2018YFA0208600)的支持🧛🏻♀️。
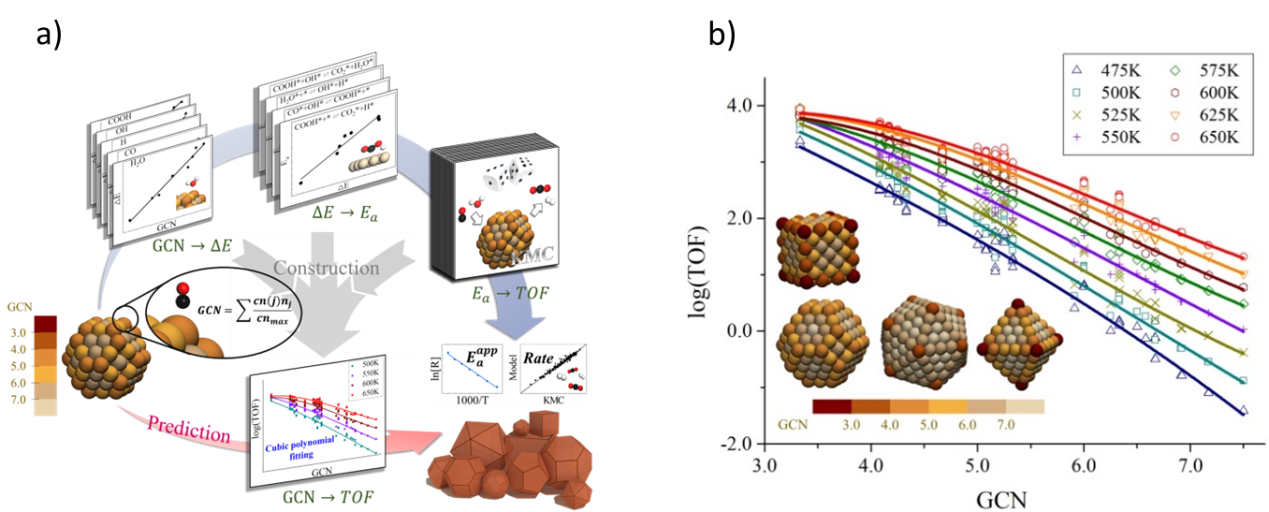
圖1 a)構建“GCN-活性”關系示意圖 b)三次多項式擬合納米顆粒的“GCN-活性”關系
全文鏈接🕵🏻:https://onlinelibrary.wiley.com/doi/10.1002/anie.202109942


