信息中心



沐鸣开户金國新教授課題組近年來一直致力於開發半夾心銥🫶🏼、銠化合物在構築具有新型拓撲結構有機金屬大環的新策略💁🏼,以及充分利用其結構特點來探索上述化合物在小分子高效識別與選擇性分離和化學信號刺激響應下的結構轉化等方面取得了一系列的研究成果(J. Am. Chem. Soc. 2020, 142, 18946-18954; J. Am. Chem. Soc. 2020, 142, 8532-8538; J. Am. Chem. Soc. 2020, 142, 13667-13671; Natl. Sci. Rev. 2020, 7, 1548-1556; J. Am. Chem. Soc. 2021, 143, 1119-1125; J. Am. Chem. Soc. 2021, 143, 5099-5105; Angew. Chem. Int. Ed. 2021, DOI: 10.1002/anie.202103264),此外,課題組還被Chemical Reviews邀請撰寫綜述文章來總結相關分子結類化合物的合成方法🕧👨🎓,同時該文章還被作為封面文章發表(Chem. Rev. 2020, 120, 6288-6325)。
1.構築新型有機金屬大環化合物的新策略——從簡單到復雜
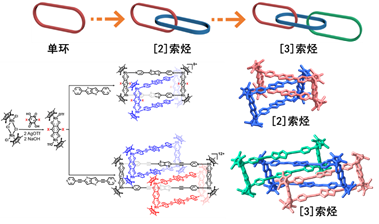
圖1-1 分子互鎖結構的高效合成
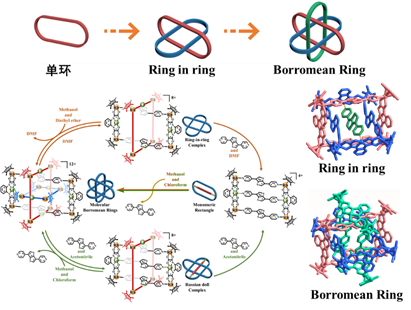
圖1-2 穿插分子與結構轉化
分子互鎖與穿插化合物的可控合成。分子索烴化合物的合理設計與選擇性合成是超分子化學領域中的熱點科學問題。它是指多個分子環通過各種分子間弱作用力相互穿插、扣鎖的分子結構。這類特殊的分子結構十分具有藝術感,同時在拓撲學上具有重要的意義。面對分子互鎖與穿插類化合物的可控設計與合成的挑戰,我們通過選擇雙核半夾心結構組裝基元與不同長度的強給電子配體,發現可以高效地合成得到一系列分子互鎖與穿插化合物(如圖1-1和圖1-2所示)👆🏽,同時上述化合物在不同溶劑以及濃度下也存在相互結構的轉化,而分子結構的深入分析也發現非共價作用力,例如氫鍵、芳環堆積作用等,在穩定這類互鎖或穿插類化合物方面扮演著重要的角色。這一研究成果發表在National Science Review上 (Natl. Sci. Rev. 2020, 7, 1548-1556)👩💻😉。
新型分子結的可控合成。分子結類化合物不僅廣泛存在於日常的生活中,同時也存在於自然界生命體中,而對於其形成的原因以及發揮作用機理的研究也逐漸吸引越來越多的合成化學家們巧妙地利用化學原理來實現人工合成分子結類化合物的可控合成🕷。金國新課題組近年來通過調節雙吡啶配體的柔性結合不同長度的雙核半夾心結構組裝基元來實現了41結,818結以及手性Solomon結的選擇性合成🤽🏼♂️。
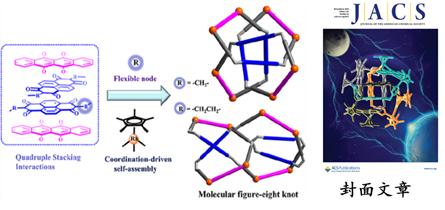
圖1-3 分子41結的選擇性構築
研究人員發現利用四重堆積作用來實現41結的構築,即通過四個組合的設計:萘二酰亞胺(NDI)基吡啶基配體和大共軛平面的半夾心金屬銠基元🫴🏻,一步法合成了41結,並且發現可以通過調節濃度以及主客體的作用來實現41結跟簡單四核大環之間的結構轉換(如圖1-3所示)。這一研究成果以全文的形式被選為封面文章發表在Journal of the American Chemical Society上(J. Am. Chem. Soc. 2020, 142, 18946-18954)👲🏿。
另外,當研究人員利用π-π相互作用作為驅動力🐕,選擇合適角度與位阻的剛性吡啶基配體,可以實現818分子結和Borromean環的構築(如圖1-4所示)👍🏿。其中818分子結是包含了500多個非氫原子的化合物🫱🏿,該結構的合成一直是復雜拓撲結構的挑戰,另外當改變吡啶基配體上的位阻來破壞它與半夾心銠有機金屬基元的π-π相互作用時,就只能得到單環化合物,這更加印證了π-π相互作用在構築818分子結和Borromean環中起到的作用,相關化合物的成功合成將為探索更加復雜的拓撲結構的合成提供重要參考。這一研究成果以全文的形式發表在Journal of the American Chemical Society上(J. Am. Chem. Soc. 2021, 143, 1119−1125)🚵🏼♂️。
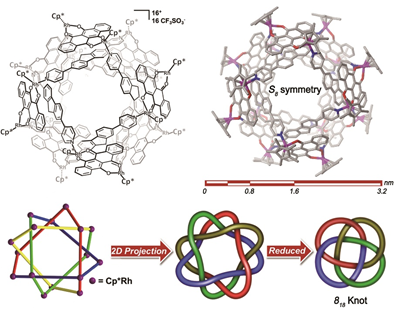
圖1-4 分子818結的選擇性構築
除此之外,Solomon結所具有的拓撲手性也引起了該課題組的關註。對於兩個非定向分子環🦬🎇,它們相互穿插互鎖一次形成的分子Hopf Links([2]索烴)不具有拓撲手性𓀆,而它們雙重互鎖形成的分子Solomon Link就具有了拓撲手性(有一對拓撲對映體P和M)(如圖1-5a)。目前合成的Solomon link的結構大多是一對外消旋體。單一拓撲手性Solomon Links的立體選擇性合成仍是一個挑戰。研究人員利用軸手性配體R-L和S-L與雙核銥化合物Ir-B(OTf)2(長度8.0 Å🦸🏽♀️,大約為2個π-π相互作用的距離)通過配位驅動自組裝的方法🚢,分別非對映選擇性的得到了分子Solomon Links的拓撲對映異構體P(Ir-1P)和M(Ir-1M)(如圖1-5b)。
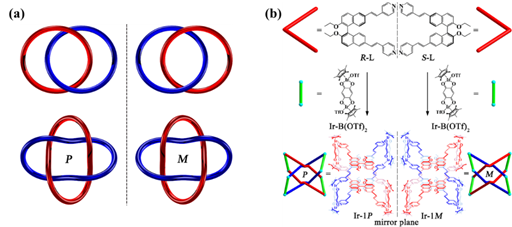
圖1-5 手性Solomon結的可控合成
此外👨🏼🔧🏄🏽♂️,課題組還被Chemical Reviews邀請撰寫綜述文章來總結相關分子結類化合物的合成方法,同時該文章還被作為封面文章發表(Chem. Rev. 2020, 120, 6288-6325🧏🏻♂️,如圖1-6所示)。
圖1-6 Chemical Reviews綜述👴🏿:配位導向分子結的構築
2.有機金屬大環化合物的性質探索——從結構轉化到應用探索
對於生命系統來說⤵️,要實現特定的生物功能🧑🏻🦱,分子尺度上的刺激響應結構轉變是常見的,並且也是必不可少的。對這種刺激響應行為的深入研究可以闡明生物學功能的基礎,也可以為設計具有刺激響應特性的分子機器提供重要的建議,而我們課題組在發展有機金屬大環的構築方法學的同時🕰,也在探索基於大環本身的應用探索,主要從拓撲結構的轉化,小分子的選擇性識別與分離以及超分子作用力導向的對位碳硼烷B-H鍵選擇性活化三個方面出發🤹🏽♀️。

圖2-1 硫醚可控氧化實現的分子Borromean環的刺激響應性拓撲轉化
化學刺激響應下的拓撲結構轉化。2020年的Springer Nature旗下期刊《通訊 化學》提出了化學領域亟待解決的十三個問題之中,對於分子結功能化的探索則被列為第一位,而我們課題組也在一直在探索對所得到的基於有機金屬大環的分子穿插互鎖或分子結這類化合物的應用,近日,金國新教授課題組將硫醚化合物的可控氧化發展成為一種結構後修飾的方法🤙🏽🧗🏼♂️,利用含有硫醚單元的雙吡啶配體與多齒配體在金屬角的作用下構築成一系列超分子有機金屬化合物🍕,之後通過精確控製氧化劑的加入量來實現超分子結構中硫醚配體的可控氧化🧏🏼♀️👚,不但實現了由雙核有機金屬大環到四核有機金屬大環化合物的刺激響應性超分子結構轉化,而且也實現了由分子Borromean環到四核有機金屬大環化合物的刺激響應性拓撲轉化(如圖2-1所示)🏋🏿,這一成果被選為內封面發表在Angewandte Chemie International Edition上 (Angew. Chem. Int. Ed. 2021, DOI: 10.1002/anie.202103264)。
小分子的選擇性可控捕獲與高效分離。有機金屬半夾心銥🙊、銠的框架材料,因其具有可調控的分子空腔,以及良好的溶解性,可以選擇性地結合客體分子Ⓜ️,這對理解客體結合過程中的潛在機理💆🏻、熱力學及特殊作用力因素方面至關重要。在前期的研究探索中,研究人員發現基於有機金屬的大環或籠狀化合物對於一些小分子具有很好的主客體化學性質,同時可以通過獲得單晶結構來深入探索其形成的驅動力,但是如何做到小分子的特異性識別的同時也能做到可控釋放則成為金國新教授課題組一直以來的探索目標。
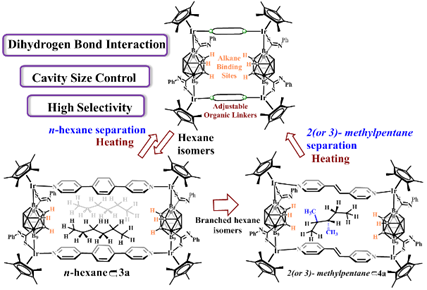
圖2-2 雙氫鍵的相互作用用於識別於分離烷烴
金國新課題組將碳硼烷構築單元引入到有機金屬環狀化合物當中,實現了不同位置的B-H鍵活化。由於碳硼烷獨特的化學特性,金國新教授課題組對碳硼烷構築的有機金屬環狀化合物的應用價值做了探索👨👧👦。因為B-H鍵中H (2.20)原子的電負性要比B (2.04)原子高,所以B-H鍵被極化為Bδ+-Hδ−的形式,因此它是有可能與Xδ−-Hδ+ (X = O (3.44), N (3.04), S (2.58), C (2.55))單元形成雙氫鍵的相互作用。受這種雙氫鍵作用的啟發,我們利用碳硼烷構築的超分子化合物實現了烷烴化合物的選擇性分離(如圖2-2所示),並且通過單晶結構合理的解釋了這種作用。通過設置對照實驗♠️,我們證明了雙氫鍵的相互作用在識別與分離烷烴中起到了重要作用。這一發現將推動對超分子化合物主客體化學在烷烴異構體的分離的應用🍹。這一研究成果以全文的形式發表在Journal of the American Chemical Society上(J. Am. Chem. Soc. 2020, 142, 8532-8538)👍🏽。
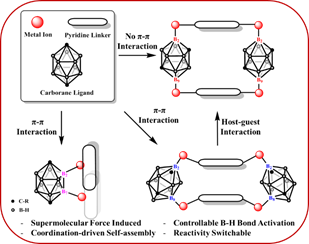
圖2-3 超分子作用力導向的對位碳硼烷B-H鍵選擇性活化
超分子作用力導向的對位碳硼烷B-H鍵選擇性活化。碳硼烷B-H鍵選擇性活化一直是碳硼烷化學關鍵的科學問題之一。然而相對於鄰位和間位碳硼烷,對位碳硼烷的活化更加困難⛹🏿♀️。這是因為對位碳硼烷結構的對稱性使它的10個硼原子化學環境相同🪆,沒有有利於對其進行親電或者親核進攻的位點🐩。所以單一位點活化的對位碳硼烷衍生物可以實現,增加活化位點就會導致多種異構體的產生,例如雙位點的活化就會導致B(2,3); B(2,4); B(2,9); B(2,8); B(2,7)五種異構體的產生。這個科學難題自對位碳硼烷發現以來一直沒有得到解決🀄️。研究人員在這篇工作中描述了如何使用超分子作用力來實現對位碳硼烷選擇性B-H鍵活化的策略🆙。在這種策略中🐘,B(2,8)-H或B(2,7)-H活化可以利用吡啶配體之間的π-π相互作用來誘導實現🧠。而在有機金屬籠狀化合物當中,通過主-客體相互作用可以避免B(2,8)-H鍵的活化🖲,進而導致B(2,9)-H鍵的活化🏵🦸🏿。吡啶配體之間的空間位阻也有利於B(2,8)-H鍵的選擇性活化(如圖2-3所示)💅。在這項工作中,研究人員證明了超分子作用力可以成為一個有效的驅動力🧏♂️,來誘導碳硼烷的B-H鍵活化🙅🫷🏿,這將在這一領域開辟出新的機遇。這一研究成果以全文的形式發表在Journal of the American Chemical Society上(J. Am. Chem. Soc.2021, 143, 5099–5105)🈲。
以上工作得到了國家自然科學基金、聚合物分子工程國家重點實驗室以及上海市分子催化和功能材料重點實驗室的資助🦵🏽。

訪問課題組主頁獲取更多研究成果(http://www.jingroup.fudan.edu.cn)👴🏽。


